Atomic simulations passed through a MLCG model
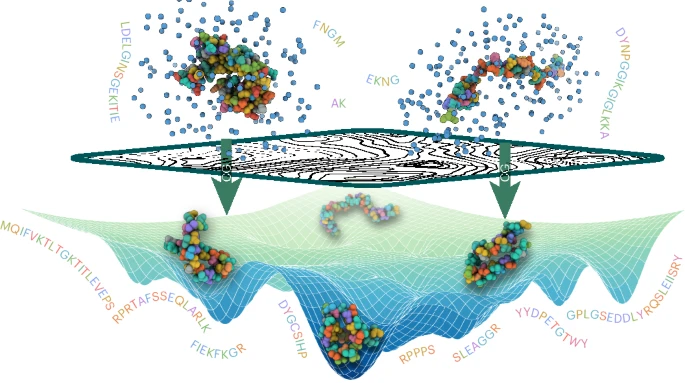
Atomistic simulations of various proteins with various sequences and secondary structure arrangements are processed through a machine-learned coarse-grained model.
Once trained, this model is able to run efficient simulations and explore the conformational landscape of any sequence.
Reference
Conceptual overview of MLCG
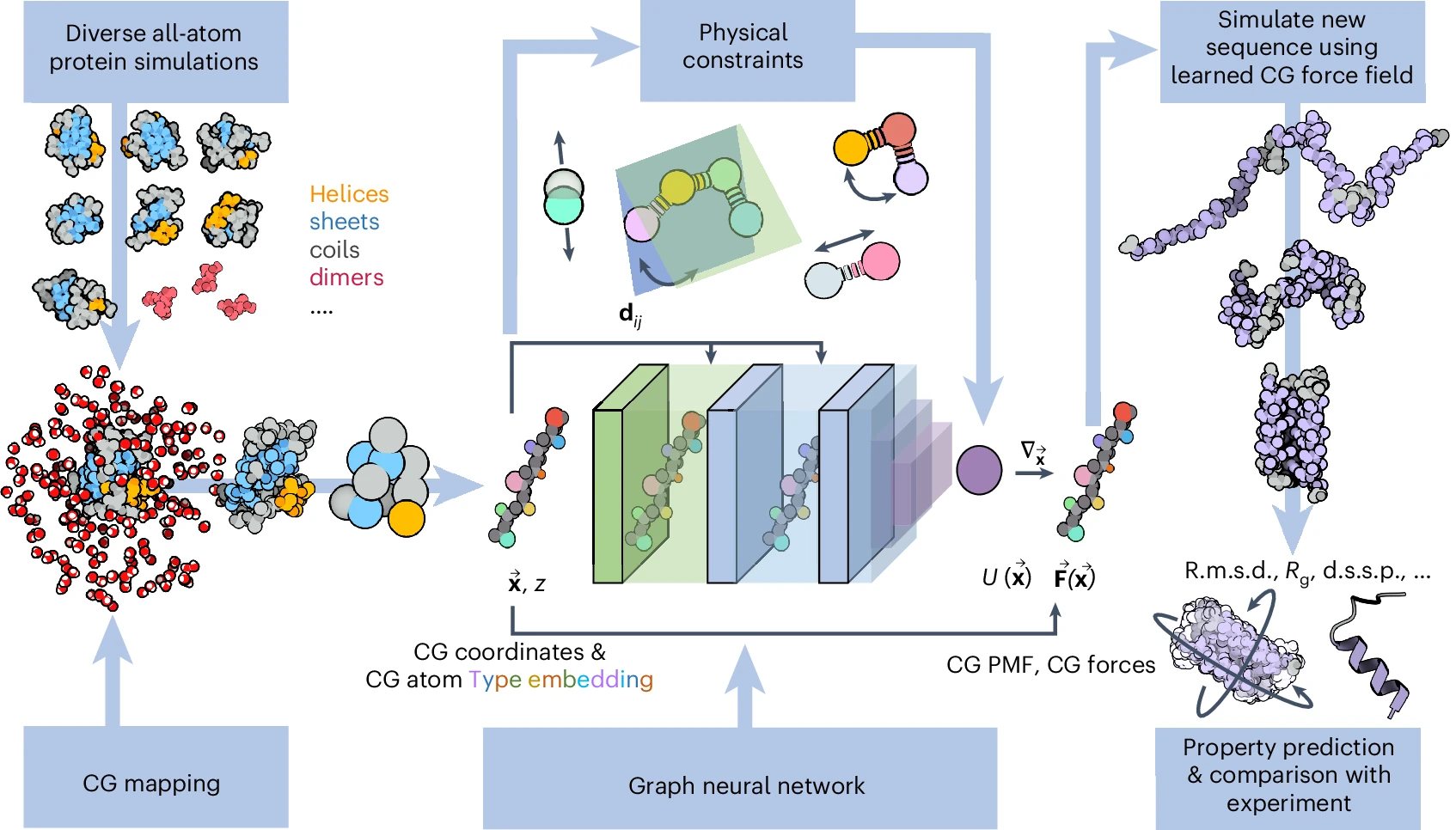
Pipeline for building and testing a transferable, bottom–up, machine-learned, CG protein force field from a diverse dataset of
all-atom simulations, a chosen CG resolution, and a set of basic physical prior energy terms (bonds, angles, dihedrals and purely repulsive interactions).
The CG atom types z and CG coordinates x are transformed into pairwise distances dij are fed into the neural network architecture to predict the CG effective potential
energy U and corresponding CG forces F. The trained neural network can subsequently be used to simulate new sequences and predict observables
such as root mean square deviations (RMSD), radii of gyration (Rg) or dictionaries of secondary structure in proteins (d.s.s.p.).
Reference
Conceptual overview of RANGE
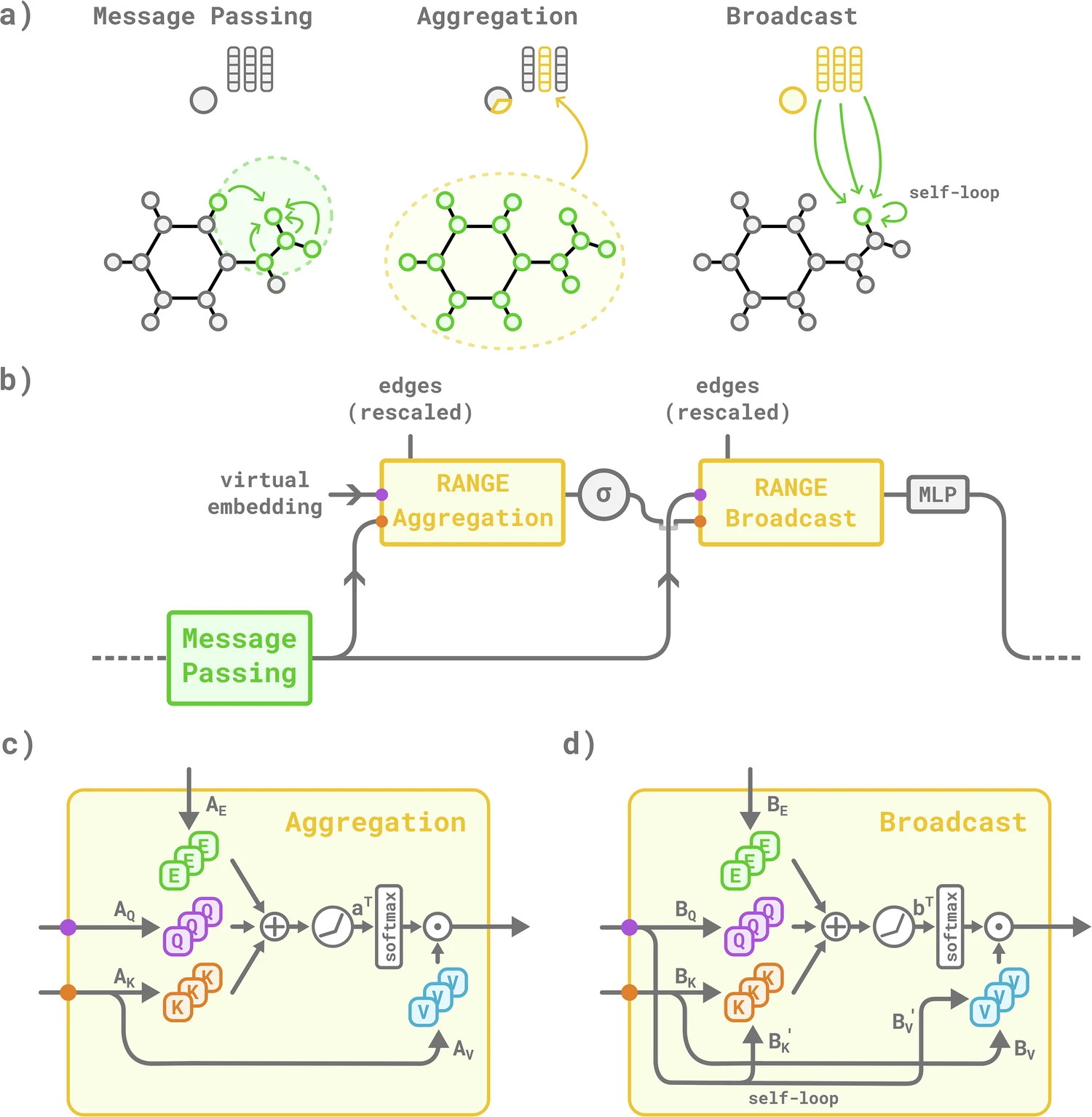
In (c) and (d), the aggregation and broadcast blocks project senders and receivers onto key and query spaces, respectively, via the linear layers AK, AQ and BK, BQ. A positional encoding projected onto the edge space via AE and BE is included in the calculation of the attention weights. During the broadcast phase (d), a memory effect, modeled by self-loops, is introduced for balancing local and global information content inside each graph node. Lastly, the attention weights are used to modulate the contributions of the receivers projected via the AV and BV linear layers.
Reference
some of the long range tasks that RANGE can achieve
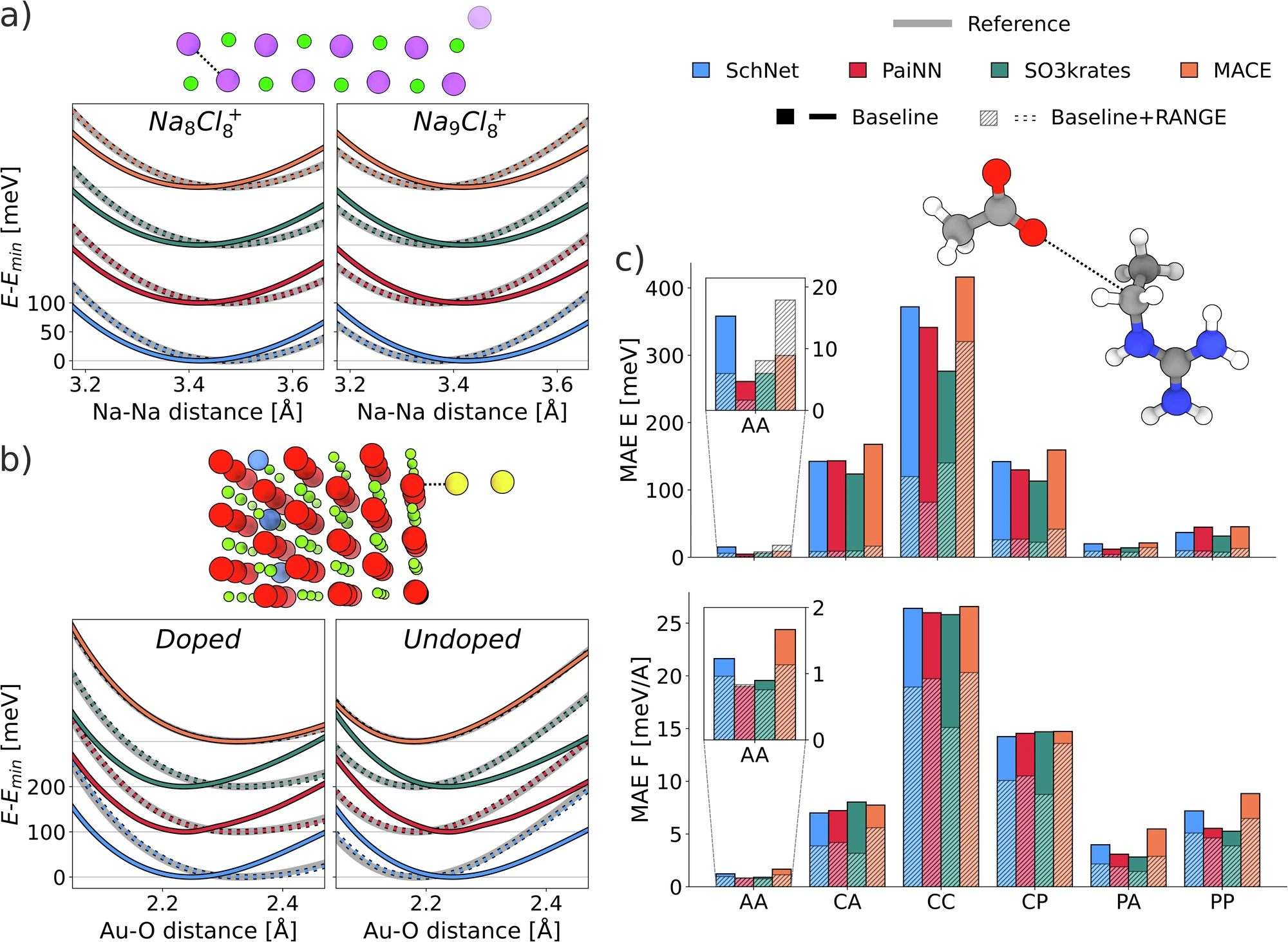
RANGE-extended message-passing neural networks (SchNet, PaiNN, SO3krates, and MACE) are compared to their baseline versions in a set of tasks designed to assess the ability to model long-range effects. (a) Relative energies of Na and Na⁺ systems (Na in purple, Cl in green) as a function of Na–Na distance, as highlighted in the structure figure. As the furthest Na atom is added or removed, the charge is kept constant. Offsets are added to distinguish the different models. (b) Relative energy of periodic MgO as the Au₂ dimer approaches the surface of the Al-doped and undoped crystal (Mg in red, O in green, Al in blue, and Au in yellow) as a function of the Au–O distance, as highlighted in the structure figure. Offsets are added to distinguish the different models. Structures and reference energy in (a) and (b) are taken from48. (c) Mean absolute error (MAE) of energy and forces of different organic dimers in an extrapolation task beyond the distance cutoff explored during training. Results are divided by the electronic distribution of the molecules in the dimers: (A)polar, (P)olar, (C)harged. The depicted molecule is extracted from the CC test set. The reported legend is shared among all panels. Source data are provided as a Source Data file.
Reference
Learning data-efficient coarse-grained molecular dynamics from forces and noise
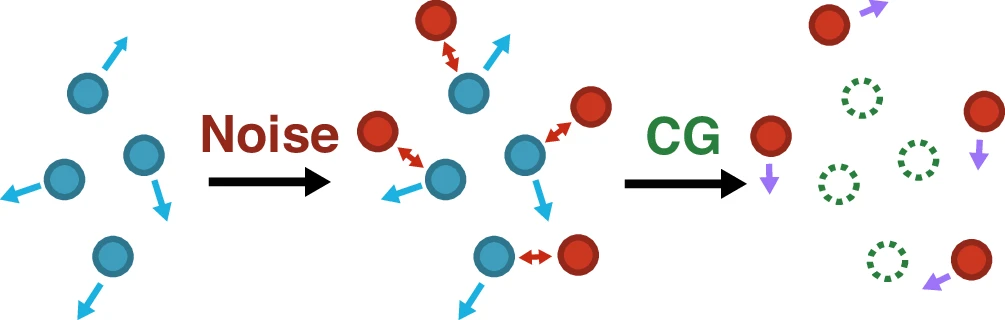
Training data consisting of “real” particles with associated forces (blue) are first combined with noise to add additional sites (red); these new particles interact with the original particles, changing forces throughout the system. The “real” particles (green dotted circles) are then systematically CG out and associated with a linear combination of “real” and noise-derived force information (purple), providing data for subsequent force-field training.
Reference
Breaking the Barriers of Molecular Dynamics With Deep-Learning: Opportunities, Pitfalls, and How to Navigate Them
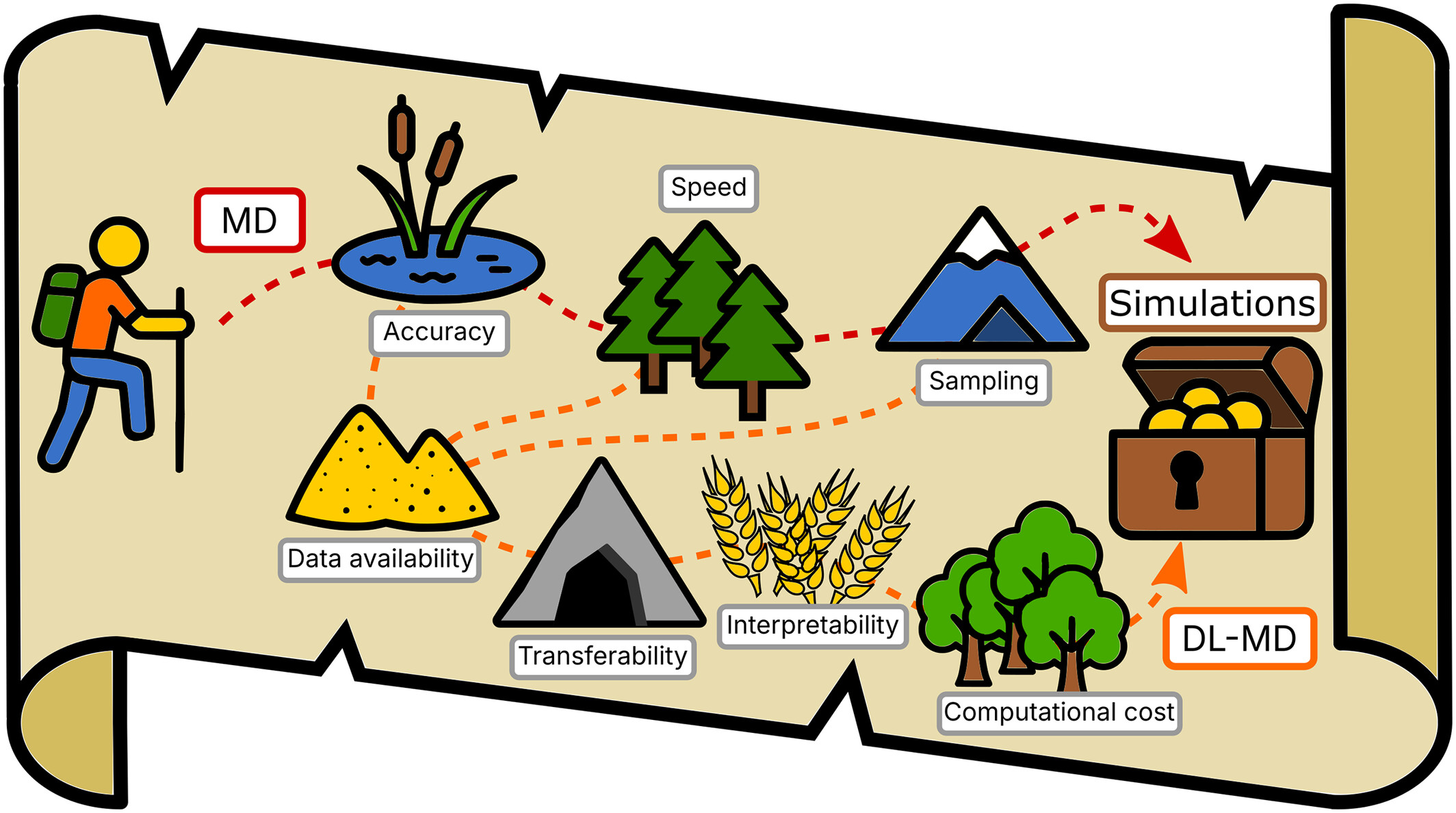
The scientist is hiking towards the treasure of accurate and predictive simulations of relevant phenomena. Molecular Dynamics shows a path riddled with obstacles such as accuracy, speed or sampling issues. Deep-learning offers a way around these obstacles, but runs into hurdles of its own.
Reference
Peering inside the black box by learning the relevance of many-body functions in neural network potentials
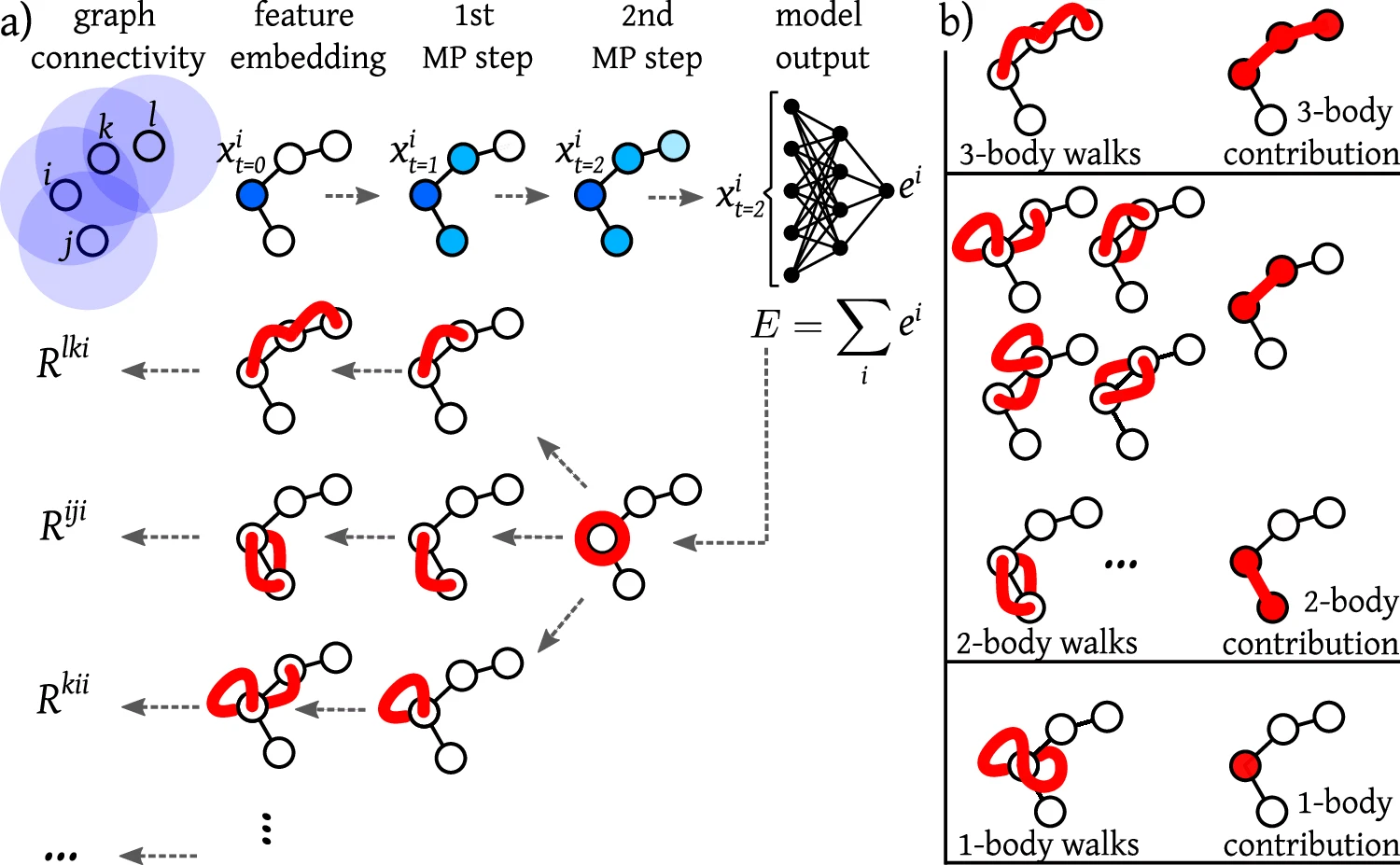
(a) In Graph Neural Networks, the input graph is defined by a cutoff radius (illustrated in shaded blue) that determines the direct neighbors for each input node. By iterative message-passing (MP) steps in multiple layers (layer index t ∈ {0, 1, 2}), information can be exchanged between more distant nodes (outside of the cutoff region), updating the representation of each node. The model output (in our case the potential energy E) is obtained by passing the learned feature representations through a multilayer perceptron and final pooling over the individual bead energies ei. Obtaining the relevance Rwalk involves propagating the output back through the network, by considering the connections between each node in one layer and the nodes in the previous layer. This procedure defines “walks” across the network layers. b The walks involving the same subset of n nodes are aggregated to obtain a decomposition of the output into n-body contributions.
Reference




